
Welcome to the PPI-MASS user guide! Here you will find instructions for the common tasks of this platform, such as submitting jobs and checking the results. You can find more information about the server and its implementation in the article published in Frontiers in Molecular Biosciences.
PPI-MASS is a web platform for the post-processing of mass-spectrometry (MS)-based proteomics data. Specifically, PPI-MASS gathers relevant information from different databases about proteins associated to a target protein, which were detected through MS experiments. Using that information, the user can identify putative protein-protein interactions to be studied in a pharmacological context. The gathered information includes:
Based on that information, the user can filter out a protein set according to different fields of interest in order to identify putative candidates to establish physical association with the target protein.
To submit a job, you will have to fill out the form found in the submit job section.
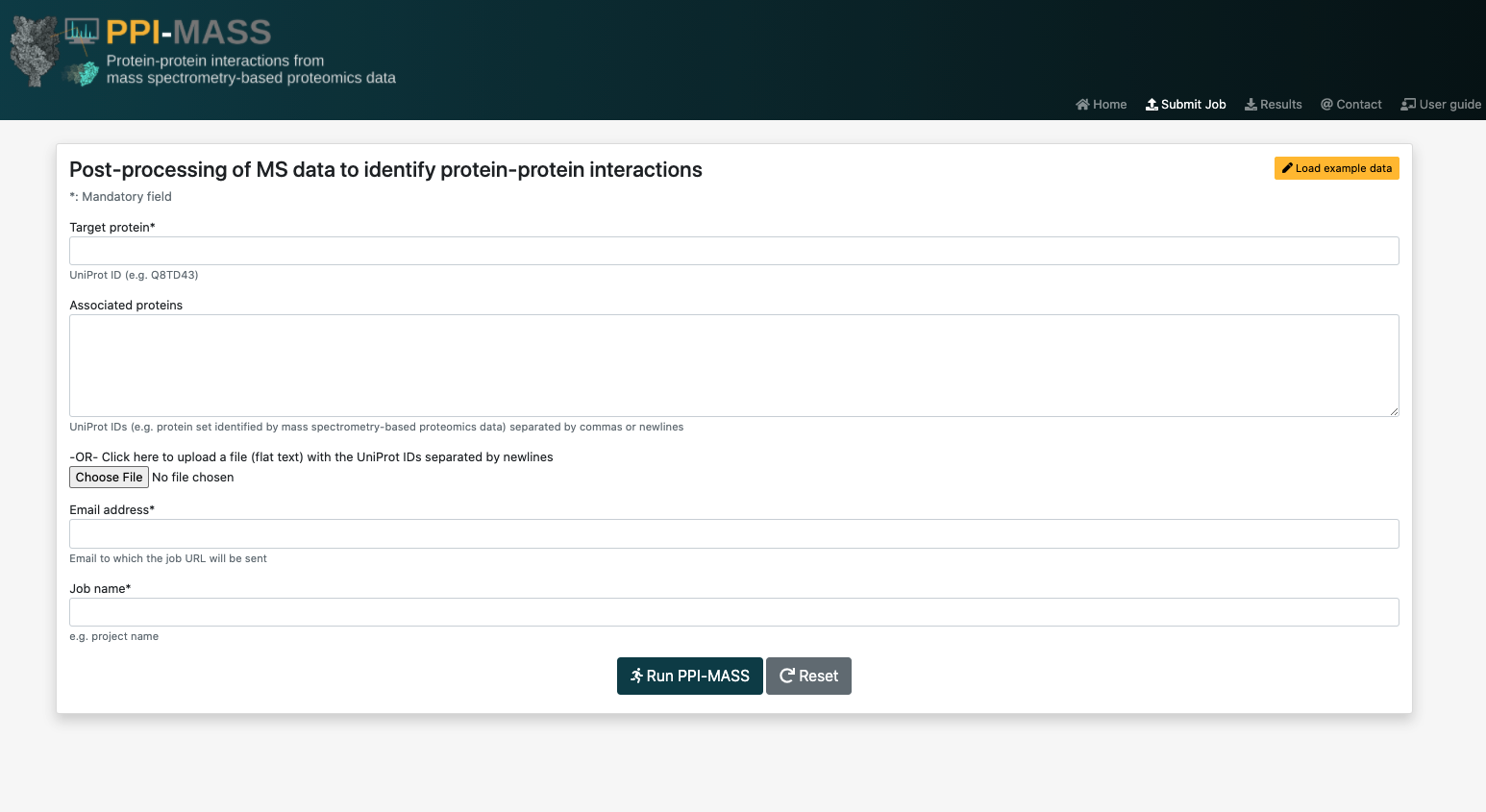
The list of proteins to be analyzed by the PPI-MASS server may submitted to the platform either by typing the UniProt accession codes into the text field separated by commas or newlines, or by uploading a plain text file with the accession codes separated by newlines. An easy way of generating this file is by copying and pasting the cells of an Excel file directly into a plain text file.
Take into account that when you select a file to upload, the contents of the "Associated proteins" field will be ignored. That is, the protein list in the uploaded file will take priority over the protein list in the field.
After submitting your job, you will be redirected to the results page, where you will be able to monitor the job's progress.
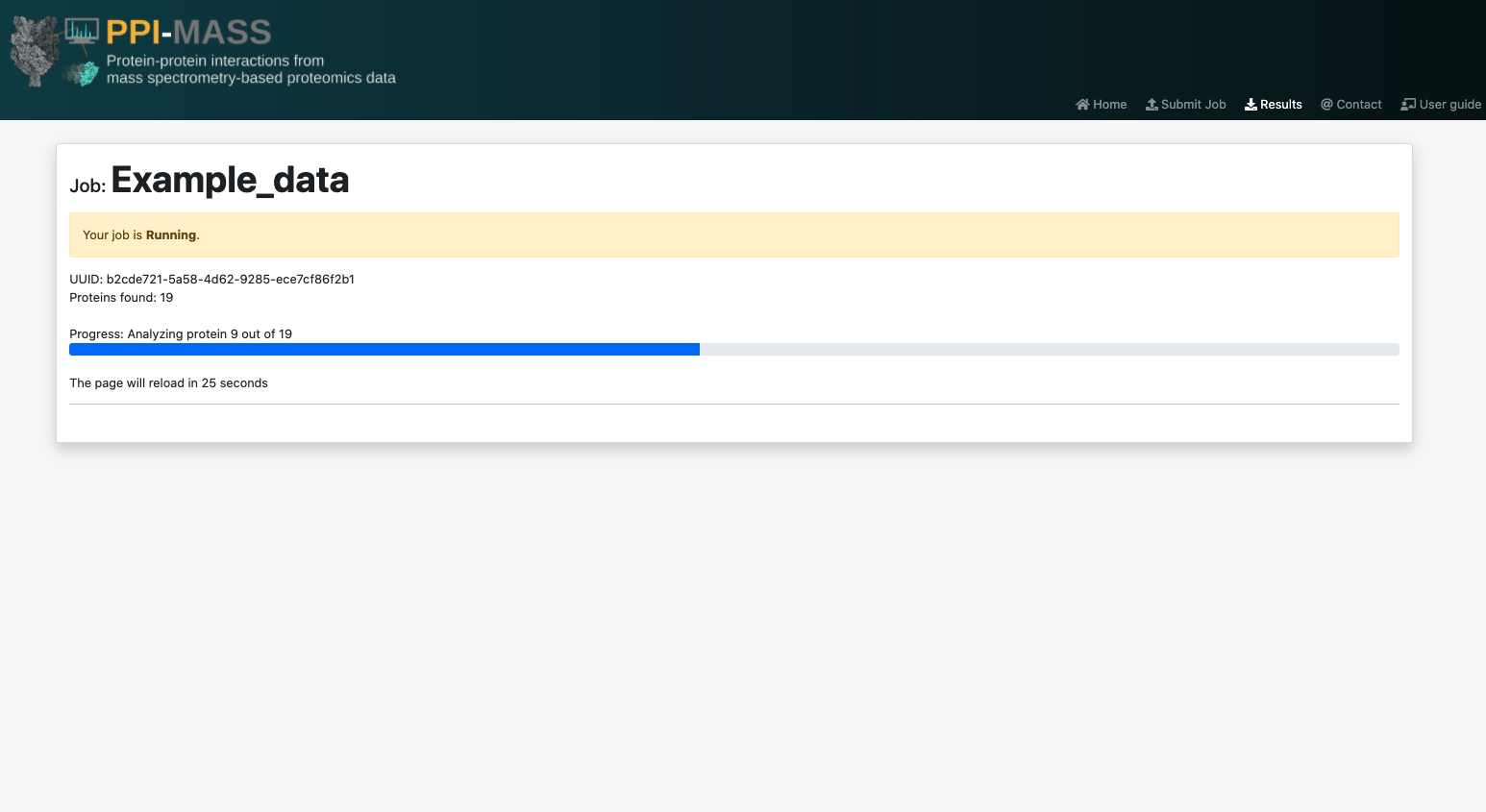
Once your job is executed, you will see the UUID job (which can be used to search for your job in the search page). It will also report the amount of proteins analyzed, the amount of proteins for which no information was found (specifying the accesion codes). It should be noted that this information is a result of the job's pre-processing stage, so it might take a while to appear. The results page will automatically reload to update the job's progress every 30 seconds until the results are ready.
Alternatively, you will also receive an e-mail with a link to access the results any time you wish. However, it is strongly suggested to save the job's URL or UUID.
Any UniProt accession codes that are not found in our registries will be ignored and indicated in the job's results page. This might be due to the codes being invalid, obsolete or due to our registries not being up to date. Therefore, they may require to be double-checked manually.
You can access the job's results directly through the link sent via e-mail or by introducing the UUID into the search box in the search page.
Using PPI-MASS you will be able to get the following information for each protein included in the data set:
Once your job is finished, you will see an interactive table with the all the available information about the submitted proteins. Here you will be able to filter out the protein set based on different fields of interest. Fields that usually present a lot of information (such as Biological process or the Associated pathologies) will show "N/A" if there is no information available or "YES" if there is, in which case you will be able to see the full results by pressing the "Show more" ( ) button.
On the other hand, the "Go to..." ( ) button contains a hyperlink to the external source from which the information in that field was obtained. In the case of the Associated drugs field, such hyperlinks can be found within the corresponding modal window that opens after pressing the "Show more" button.

Additionally, with the export ( ) buttons, you will be able to export the results in Excel, CSV and PDF formats, respectively. Any applied filters will be reflected in the exported files.
Filters can help you to reduce the number of proteins that you are working with. All filters can be applied simultaneously and the table will automatically update accordingly with the proteins with matching values. There are two main types of filters: checkboxes and text fields.
Checkboxes are used to filter out proteins based on structure availability. By default, all proteins are visible, but you can choose to only show those with PDB structures available, or proteins that only have a model in Swiss-Model, or proteins that don't have available 3D structures. These three filters can be combined in any way you want.
The text fields allow you to search for text within each column. They are case insensitive and can find partial matches as well, that is, they will match with incomplete words. In the case of the fields that have hidden text within modal windows (such as the tissue specificity field), the filter will search for text inside those windows too. You may also simply type "YES" into these filters to search for proteins with information available inside that particular field. You may introduce more than one pattern into these fields, all of which will be required to appear in the field for it to be considered a match. For instance, the string "P07 4" will match the accession codes that have P07 and also a 4 somewhere in them.
The text fields also allow the use of regular expressions for advanced pattern searching. For instance, the pattern "^P.*7$" in the UniProtKB entry filter will find all proteins whose accession code starts with P and ends with 7, with any number of characters in between.
Finally, if you want to reset the table to its initial state, you can simply press the "Clear filters" ( ) button.